

VAMOS ENTENDER OS PRÍONS E SUAS CONDIÇÕES PATOLÓGICAS?
Em 1982 Stanley B. Prusiner utilizou esse termo, “PRÍONS”, ao descrever condições neurodegenerativas em mamíferos e que se caracterizavam em alterações espongiformes no cérebro, conhecidas como espongiformes transmissíveis (EETs).
Esse tipo de doença ocorre devido a alterações conformacionais de uma proteína humana chamada de PrPSc que passa a se acumular no SNC. Essas doenças são de caráter lento.
Agora, vamos ver um pouco sobre as doenças de príons e suas características:
Doença de Creutzfeldt-Jakob (DCJ)

Possui quatro subtipos, sendo eles:
-> Em primeiro lugar, as epidemiologicamente consideradas esporádicas e etiologicamente consistem na alteração conformacional espontânea na proteína príon ou também por uma mutação somática do gente;
-> Em segundo lugar, a epidemiologicamente considerada familiar e sua etiologia consiste em mutações na linhagem germinativa que tornam as proteínas suscetíveis à alteração conformacional;
-> Posteriormente, a epidemiologicamente considerada iatrogênica com sua etiologia marcada por transplante de córnea e/ou dura-máter, eletrodos de ECG invasivos e outros procedimentos neurológicos.
Além disso, pode ser transmitida por administração de GH que transmitiram príons;
-> E para fechar, a epidemiologicamente considerada variada (vDCJ) e etiologicamente caracterizada por ingestão de carnes bovinas contaminadas com encefalopatia espongiforme bovina (EEBV).
Doença de GErstmann-Sträussler-Scheinker (GSS)

Sua epidemiologia é a insônia familiar fatal e sua etiologia é a mutação na linhagem germinativa D178N associada ao polimorfismo 129M localizado no mesmo gene.
Há também essa doença relacionada com insônia fatal esporádica com alteração conformacional espontânea na proteína do príons ou também uma mutação somática do gene.
Doença Kuru:
É uma doença epidemiologicamente considerada de transmissão entre pessoas e etiologicamente marcada por proteínas de príon, transmitida por canibalismo ritual dos indígenas de Papua-Nova Guiné.
PARA FACILITAR A COMPREENSÃO VAMOS DIFERENCIAR OS PRÍONS DAS PARTÍCULAS VIRAIS EM RELAÇÃO AOS PRÍONS
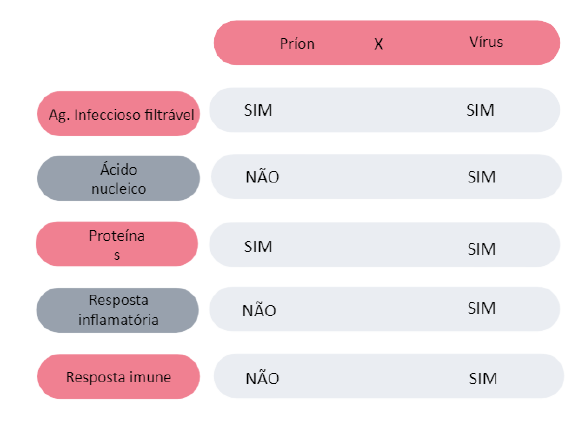
Além dessas características inseridas na tabela podemos citar:
- Morfologia definida apenas em vírus;
- Proteases (enzima de desinfecção): alguns vírus e nenhum príon;
- Efeito citopatológico: apenas nos vírus;
- Período de incubação: variável no vírus e longo no príon;
- Produção de interferon: apenas nos vírus.
Agora que você já conhece as doenças e o que são príons, vamos compreender sua patogênese
A doença relacionada com príons acontece quando uma dobra anormal da proteína PrPSc ocorre e, na maioria das vezes, não é solúvel e resistente à proteases.
Dessa forma, esse príon consegue escapar do sistema imunológico e se acumular no cérebro. O cromossomo dos humanos que portam esse gene PrP é o número 20.
Vamos entender a formação da PrPSc
- Endógena: mutação somática esporádica do gene PrP, ou alteração conformacional espontânea da PrPc normal; Mutação do gene em questão na linhagem germinativa – A PrPc é considerada instável e se converte com facilidade em PrPSc.
- Exógena: doenças priônicas infecciosas. Podem ser transmitidas entre animais e humanos.
Depois de formada, a PrPSc vai interagir com a PrPc e catalisar a conversão desta em PrPSc. Dessa forma, há uma reação de cadeia chamada de “replicação do príon”.
Em relação a encefalopatia espongiforme, tem-se a formação de neurônios vacuolizados (por isso o nome espongiforme) e a perda da sua função. Isso se dá devido à ausência da resposta imune e, posteriormente, à inflamação.
É possível observar ainda a presença de células gliais adjacentes, proliferação de astrócitos, hipertrofia, placas de fibrilas e amiloides.
A PrPSc será captada pelos neurônios e fagocitada por células fagocitárias e sua degradação não será facilitada, proporcionando a característica espongiforme da doença no tecido cerebral.
Entendendo as manifestações clínicas das doenças causadas por príons
- DCJ Esporádica: pacientes apresentam demência, perda de equilíbrio (ataxia), espasmos musculares (mioclono), atividade periódica de alta frequência evidenciada no EEG (eletrocefalograma). Também é possível notar fadiga, cefaléia, vertigem, problemas comportamentais e alterações do sono. Com prognóstico ruim o paciente pode ficar acamado e sem conseguir falar;
- DCJ familiar: acomete pessoas na faixa etária de 40 a 60 anos ou também aos 25 anos de idade.
- vDCJ: alterações comportamentais na fase inicial da doença e algumas sensações de dor. Além disso, também apresentam demências com duração da doença com média de 14 meses;
- GSS: Disartria, ataxia e demência são os principais sintomas;
- IFF: insônia sem possibilidade de tratamento, disautonomias (pressão arterial alterada, temperatura, frequência cardíaca e respiratória também modificadas), ataxia.
Você consegue explicar o diagnóstico dessas doenças priogênicas?
A princípio, o médico se baseia na somatória dos sintomas e nos sinais clínicos que o paciente apresenta.
Também há a exclusão de outras condições clínicas devido aos resultados apresentados nos exames (prática eliminatória).
Algumas condições devem ser analisadas com cautelas como por exemplo o EEG positivo, história familiar da doença, mutações na linhagem germinativa em casos familiares e práticas alimentares e o saneamento básico da região em que o paciente reside.
Em resumo, para o diagnóstico neuropatológico é necessário uma biópsia do cérebro com imuno-histoquímica ou immunoblot. Porém, o diagnóstico oficial se dá a partir da inoculação do material cerebral em espécies animais e, posteriormente, da observação das manifestações clínicas e patológicas desses animais.
